Histone Deacetylase Structure and Function
Reversible acetylation in the regulation of protein function is a ubiquitous posttranslational modification that rivals phosphorylation in its scope. Rooted in the discovery of histone acetylation more than 50 years ago, lysine acetylation-deacetylation cycles are now known to regulate the cellular functions of nuclear and cytoplasmic proteins in epigenetics, cell signaling, metabolism, and other biological processes. Thus, acetylation in biology is not limited solely to histone proteins in the nucleus.
Three classes of proteins are involved in the chemical biology of the acetylome: “writers”, or histone acetylases (also known as lysine acetyltransferases); “readers”, i.e., bromodomain-containing proteins that specifically recognize the acetyllysine moiety; and “erasers”, or histone deacetylases (HDACs, also known more generally as lysine deacetylases since both histone and nonhistone proteins are substrates). Our group focuses on the metal-dependent HDACs, which typically require a single zinc ion for catalytic activity in the hydrolysis of acetyllysine:

Importantly, HDACs are validated drug targets for human disease. For example, upregulated HDAC activity is associated with tumorigenesis, and HDAC inhibitors induce growth arrest, differentiation, and apoptosis of cancer cells. The “pan-HDAC” inhibitors Romidepsin, Vorinostat, Belinostat, and Panobinostat are potent inhibitors of all HDAC isozymes and are currently approved for cancer chemotherapy. However, even though the inhibition of multiple isozymes by a pan-HDAC inhibitor contributes to serious side effects, no isozyme-selective HDAC inhibitors are currently approved as drugs. The development of HDAC-selective inhibitors thus addresses a critical unmet need in the clinic. Notably, HDACs may also serve as drug targets for other diseases, including immune disorders, neurodegenerative diseases, and peripheral neuropathy. Given the significance of the HDACs for the US public health, this metalloenzyme family provides a unique platform for chemical, biological, and medical innovation.
Our group is pursuing X-ray crystallographic studies of HDAC6, HDAC8, and HDAC10. Our work with HDAC8 focuses on site-specific mutants identified in children diagnosed with Cornelia de Lange Syndrome (CdLS), a congenital birth defect diagnosed in one out of 10,000 births. Structural analysis of CdLS HDAC8 mutants allows us to understand the molecular basis for compromised catalysis, which leads to compromised cohesin function during the cell cycle. Our work with HDAC6 focuses on the structural basis of substrate specificity in each catalytic domain:

Y. Hai, D. W. Christianson, Nat. Chem. Biol. 2016, 12, 741–7.
Since catalytic domain 2 (CD2) of HDAC6 serves as the tubulin deacetylase, this domain is the target of isozyme-specific inhibitors that can be used in cancer chemotherapy. Inhibition of HDAC6 CD2 suppresses microtubule dynamics, which leads to cell cycle arrest, apoptosis, and cell death.
Finally, our structural and enzymological studies of HDAC10 led to the discovery that this isozyme is a polyamine deacetylase (PDAC). HDAC10 is a two-domain protein in which the PDAC domain is fused with a pseudo-deacetylase (ΨDAC) domain that adopts the same fold but lacks zinc binding activity and consequently lacks catalytic function. However, the ΨDAC domain is important for the stabilization of the PDAC domain in a catalytically active conformation.
 Y. Hai, S. A. Shinsky, N. J. Porter, D. W. Christianson, Nat. Commun. 2017, 8, 15368.
Y. Hai, S. A. Shinsky, N. J. Porter, D. W. Christianson, Nat. Commun. 2017, 8, 15368.
Structural basis of terpene biosynthesis
Terpenoid cyclases catalyze the most complex chemical reactions in biology, in that on average more than half of the substrate carbon atoms undergo changes in bonding and/or hybridization during the course of a single enzyme-catalyzed reaction. Even for the most sluggish terpenoid cyclase, the rate enhancement factor over the uncatalyzed rate is immeasurably large since these multi-step reactions cannot occur in the absence of a protein catalyst.
Terpenoid cyclases are unique among enzymes in that they are characterized not only by their reaction rates (i.e., traditional steady-state kinetics), but also by how many products they make: high-fidelity cyclases generate a single, exclusive product, whereas promiscuous cyclases generate multiple products, all from a single substrate. This reflects nature’s economical approach to chemical diversity, in that small evolutionary steps for a cyclase can yield dramatically different product arrays from just a single substrate. With more than 80,000 terpenoid natural products identified to date in all forms of life, the chemodiversity of terpenoid biosynthesis is staggering. Many terpenoids exhibit beneficial pharmacological properties, e.g., as antibacterial, antifungal, or anticancer agents – the blockbuster cancer chemotherapeutic drug Taxol (paclitaxel) is a well known example. Furthermore, terpenoid drugs can be generated in whole or in part through synthetic biology approaches. To add value to the toolbox of synthetic biology, we must develop a command of structure-function relationships in terpenoid cyclases as renewable and cost-effective approaches to drug development are explored.
Our group has determined the first crystal structures of class I terpenoid cyclases that catalyze the cyclization of 10-, 15-, and 20-carbon substrates. These enzymes utilize a trinuclear magnesium cluster to trigger the ionization of a prenyl diphosphate substrate, resulting information of an allylic carbocation that undergoes additional carbon-carbon bond-forming reactions. We are particularly interested in variable domain architectures found for these cyclases and how domain architecture influences catalysis. The chemistry of catalysis for a class I terpenoid cyclase occurs in the α domain, which is typically found as a single-domain protein in bacteria and fungi. In plants, however, the α domain is fused with one or two other domains so that the overall protein architecture is described as αβ or αβγ. In contrast, the chemistry of catalysis in a class II terpenoid cyclase occurs at the βγ domain interface in enzymes with βγ or αβγdomain architecture:
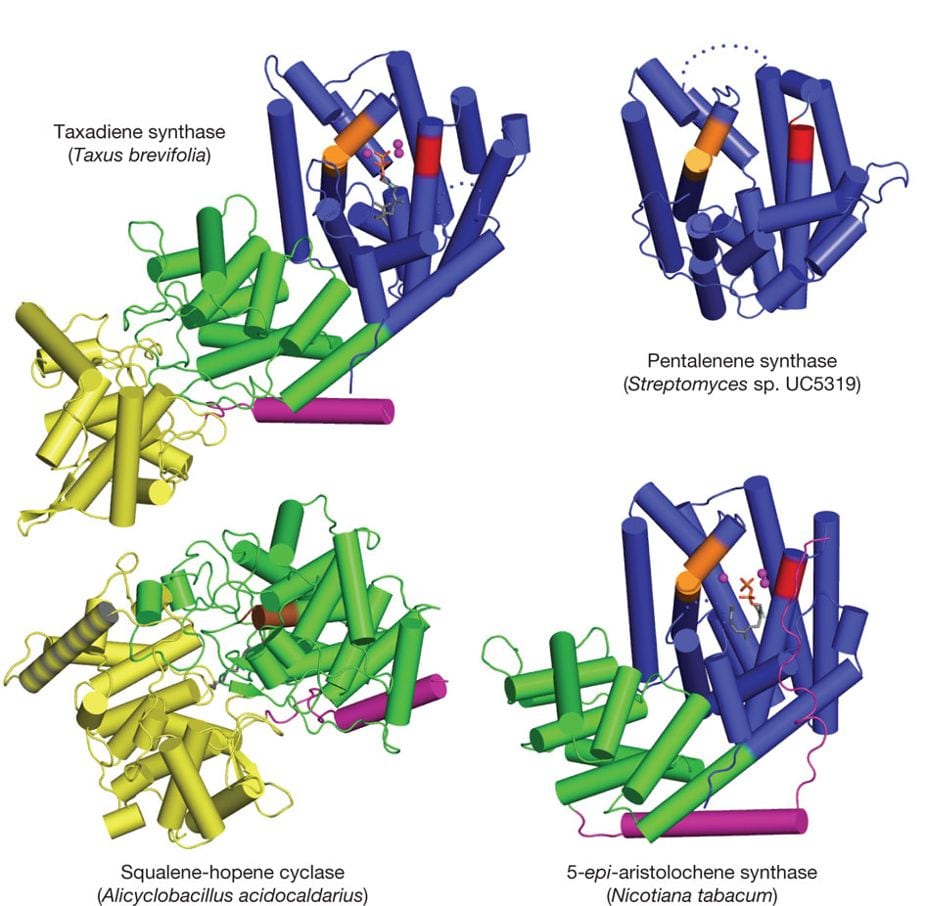 M. Köksal, Y. Jin, R. M. Coates, R. Croteau, D. W. Christianson, Nature 2011, 469, 116–20.
M. Köksal, Y. Jin, R. M. Coates, R. Croteau, D. W. Christianson, Nature 2011, 469, 116–20.
We have recently begun studies of unusual assembly-line terpenoid synthases that adopt αα domain architecture and assemble to form complex oligomeric structures. In these fungal enzymes, one α domain links 5-carbon building blocks to generate a 20- or 25-carbon linear isoprenoid diphosphate, and the second α domain catalyzes the cyclization of the isoprenoid diphosphate to generate a muti-ring product. Our enzymological studies indicate that there is channeling between active sites in the diterpene synthase fusicoccadiene synthase. To date, our X-ray crystal structures of the individual α domains of this enzyme fit into the ab initio molecular envelope of the intact hexamer calculated from small-angle X-ray scattering data provide a valuable reference point for understanding structure-mechanism relationships in this assembly-line terpenoid synthase.
 M. Chen, W. K. W. Chou, T. Toyomasu, D. E. Cane, D. W. Christianson, ACS Chem. Biol. 2016, 11, 889–99.
M. Chen, W. K. W. Chou, T. Toyomasu, D. E. Cane, D. W. Christianson, ACS Chem. Biol. 2016, 11, 889–99.