Genetic Diseases Associated with the NALCN-UNC80-UNC79 Complex
The NALCN-UNC80-UNC79 protein complex forms an ion channel that allows a small amount of sodium to “leak” into each neuron to excite the cell. At resting, potassium ions also “leak” out of the cell to “silence” the neuron. This yin and yang balance between the two ionic leaks provides a dynamic regulation of resting membrane potential and generates heterogeneity in neuronal excitability in various brain regions and spinal cord (see review). More than a dozen of ion channels carry potassium leak. Unfortunately for humans, there is only one mammalian member in the NALCN gene family and there is little genetic redundancy in the sodium leak. Variations in NALCN and UNC80 genes are now known to cause some of the most severe neurological diseases in humans.
Functional studies of genetic variations in UNC80 associated with intellectual disability
Using whole exome sequencing and linkage analysis, we and others have reported more than 20 genetic variations/mutations in the UNC80 gene associated with human diseases (see diagram below). The symptoms in the affected individuals are severe and include hypotonia, encephalopathy, lack of speech and severe intellectual disability.

Disease-associated genetic variations in the UNC80 open reading frame. Established UNC80 functional domains are indicated. References: 1. Stray-Pedersen A. et al. Am J Hum Genet (2016), 2. Shamseldin, H. E. et al. Am J Hum Genet (2016), 3. Perez, Y. et al. J Med Genet (2016), 4. Valkanas, E. et al. Am J Med Genet (2016), 5. Obeid, T. et al. Metab Brain Dis (2018), 6. He, Y. et al. Gene (2018), 7. Bramswig, N. C. et al. Hum Genet (2018), 8. Kuptanon, C. et al. Gene (2019)
For any disease-implicated genetic variant, particularly those identified using whole exome sequencing, a common challenge is to establish a causal relationship between the variation and diseases. To establish such relationships, we have studied in detail the functional consequences of some of the UNC80 variations. We use biochemistry and microscopy to examine the consequence of the variations on the expression and localization of the protein in neurons, electrophysiology to check whether such variations lead to changes in ion channel properties, and CRISPR/Cas9-mediated gene editing to test whether introduce similar genetic variations into animal models (mice) leads to changes in animal physiology/behavior. With those approaches, we found that the variations can be functionally divided into several groups.
UC80 N-terminal truncations that cause complete loss of function. Several disease-associated UNC80 variations lead to stop codons in the N-terminal end of the protein (e.g. R51*, R174* and R360*). Introducing such a stop codon (equivalent to R51* in the human mutation) to the UNC80 gene in mice almost completely abolishes the sodium leak currents in neurons, and causes severe apnea and neonatal lethality in the mice, suggesting that drastic variations in UNC80 can cause severe diseases.
UC80 variations that weaken the UNC80-UNC79 interaction. A variation in the UNC79-interacting domain (R2842Q) reduces the strength of association between UNC80 and UNC79. This variation is associated with intellectual disability, although the affected individual has less severe diseases than the ones with truncation mutations at the N-terminal.
UC80 variations that prevent the channel complex to reach dendrites. UNC80 is present throughout the neurons, including soma and dendrites. Several variations causing truncation at the C-terminal (e.g. L2586*) do not reduce the amplitudes of the sodium leak in whole neurons. However, UNC80 protein with the truncations is restricted in soma and absent in dendrites. Mice with the truncation have severe apnea and neonatal lethality. Humans with those truncations have severe hypotonia and intellectual disability, suggesting the importance of a proper regulation of dendritic resting membrane potential by the NALCN-UNC80-UNC79 complex.
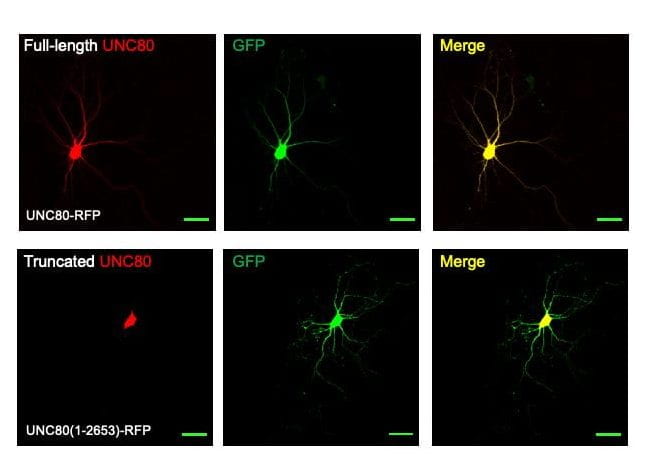
Truncations at UNC80’s C-terminal found in intellectual disability-associated diseases abolish dendritic localization. Images from cultured hippocampal neurons transfected with RFP -tagged full-length UNC80 (upper) or C-terminally truncated UNC80 (lower), together with GFP to label neuronal processes.
Diseases associated with NALCN gene variations
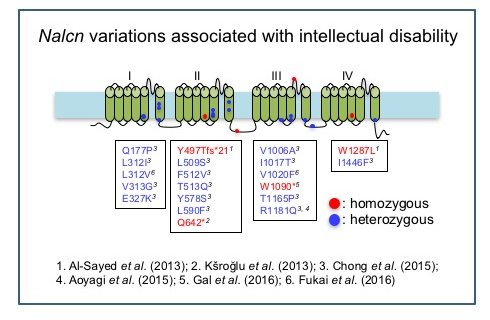
As reported in the following papers, individuals with homozygous NALCN mutations are severely hypotonic, unable to sit or stand, fail to thrive, do not develop speech, and have severe intellectual disability.
- Koroglu et al. (2013). J Med Genet; Al-Sayed et al. (2013) Am J Hum Genet; Gal et al. (2016) Eur J Med Genet.
Heterozygous de novo NALCN variations have also been found in children with severe symptoms including hypotonia, developmental delay and intellectual disability.
- Chong et al. (2015) Am J Hum Genet; Aoyagi et al. (2015) Human mutation 36, 753-757; Fukai et al. (2016) J Hum Genet.
Diseases associated with UNC79 genetic variations
Heterozygous variations in UNC79 are associated with intellectual disability and developmental delay.
Link: The Channeling Hope Foundation dedicated to NALCN-related diseases. https://www.channelinghope.org