Regulation of Neuronal Excitability by the NALCN-UNC80-UNC79 Sodium-leak Channel Complex
The brain uses electrical signals to control essentially every aspect of our life: from breathing and walking to speaking and intellectual reasoning. Hypothetically, a functional nervous system can be built based primarily on synaptic connectivity between neurons that have identical intrinsic properties (analogous to a computer memory chip built from identical transistors). In animals with a complex central nervous system, however, there is large variation in the intrinsic properties among neurons found in different brain regions or from the same area. For example, the resting membrane potential (RMP) of mammalian neurons ranges from ~ -90 mV to ~ -40 mV, depending on the types of neurons, the location and time. How this complexity in the intrinsic property of neurons is essential to the normal function of an organism is not well understood.
At resting, neurons are predominantly permeable to K+. It has been known since 1940s that neurons also “leak” Na+ into the cells. The leak leads to an RMP depolarized above the equilibrium potential of K+ (EK, ~ -90 mV). The leaks cost energy and produce heat in the brain. However, the interplay between the two leaks provides a powerful mechanism to generate complexity in RMP. The basal Na+ leak is generally very small, with a permeability (PNa) only a few percent of that K+ (PK). The small size of Na+ leak makes it possible for large fold changes in PNa/PK and large RMP dynamic ranges among neurons.
A major contributor to the basal Na+ leak is a voltage-independent, TTX-resistant, Na+ permeable cation conductance detected in many neurons. The conductance is very small, ~1% of that of the voltage-gated TTX-sensitive Na+ channels, making it hard to record. We discovered that the conductance is formed by NALCN, a voltage-independent, non-inactivating Na+– permeable (but non-selective) cation channel. NALCN is the single most dominant (~ 70% of total in the hippocampal neurons) Na+ conductance at resting. Neurons lacking NALCN are hyperpolarized by 10 to 20 mV. NALCN has similarity with the voltage-gated Ca2+ channels (CaVs) and Na+ channels (NaVs). It evolved earlier before the separation between CaVs and NaVs.
NALCN forms a complex with another two large proteins UNC80 and UNC79 (~3,000 amino acids) and a smaller one NLF-1 (FAM155A). Regulators such as neuropeptides and extracellular Ca2+ can potentiate NALCN-mediated currents by >10 fold and generate a large dynamic range of membrane potential. In humans, mutations in Nalcn and Unc80 genes lead to inability to stand or walk, lack of speech development, severe intellectual disability and premature death, suggesting fundamental physiological significance of Na+-leak mediated complexity in neuronal properties. A current focus in the lab is to understand the molecular mechanism by which NALCN complex regulates the global excitability and complexity of the brain and its contribution to animal behavior in normal and disease states.
Auxiliary subunits of the NALCN complex: UNC80 and UNC79
Ion channel pore-forming proteins usually don’t work alone. We found that NALCN, the pore-forming subunit of the channel, in mammalian brains forms a complex with two large novel proteins UNC80 and UNC79, homologs of the C. elegans Unc-80 and Unc-79, respectively. Immunodepletion of NALCN in the brain also depletes UNC80 and UNC79, suggesting that UNC80 and UNC79 are exclusively associated with NALCN and are auxiliary subunits of the Na+-leak channel complex.
NALCN, UNC80 and UNC79 are found in neuronal cell bodies, dendrites and axon, suggesting that the complex plays role in regulating local excitability of neuronal processes. Both UNC80 and UNC79 are large (~3,000 amino acids) and highly conserved, yet they do not have recognizable domains or share sequence similarity with other proteins. We are using protein chemistry, electrophysiology, mouse genetics and human disease to define protein domains responsible for inter-subunit interaction, protein localization, ionic conductance and regulation.
With two large auxiliary subunits of ~3,000 amino acids each, the NALCN-UNC80-UNC79 complex has a total of ~8,000 amino acids and is the largest known complex in the 24 transmembrane-spanning (4x6TM) superfamily of ion channels (~4,000 in CaVs and ~2,000 in NaVs). Among the 4x6TM families (NALCN, CaVs and NaVs), the NALCN-UNC80 complex also appeared to be the earliest in the evolution of eukaryotes. For example, the fungi Lichtheimia ramosa and Lichtheimia corymbifera each has two NALCN orthologs (access # CDS04857.1 and CDS14553.1 for L. ramosa) and one UNC80 (# CDS03381.1). The pore-forming α1 subunits of CaVs are also present in fungi (e.g. #CDS14339.1 in L. ramosa), but the auxiliary subunits appeared later during evolution, with the β subunit first found in choanoflagellates (a few million years later than UNC80). Similarly, NaV β subunits are not present until metazoan. Like mammalian UNC80s, the fungal UNC80 is large (2531 amino acids). Intriguingly, there is only one segment of ~800 aa that is conserved with a fragment of 769 aa of the mouse UNC80 (aa 1022-1790; 39% similarity, 21% identity). This segment is part of the NALCN-interacting domain, suggesting that functional NALCN-UNC80 interaction evolved early in the common ancestor of fungi and metazoa. Although UNC80s in both animals and fungi are large, the rest of the protein in the N- and C- termini have diverged, with a C-terminal fragment in animals evolved later in metazoa to associate with UNC79, which appeared several hundred million years later, first in sponges.
UNC80 functions like the β subunits of CaVs and NaVs. It potentiates the NALCN channel currents by at least two mechanisms: promoting the traffic of the pore-forming subunit NALCN to the cell surface to increase the number of functional channels and, perhaps, increasing channel opening probability. UNC80 is required for the regulation of NALCN-mediated Na+ leak by extracellular Ca2+ and neuropeptides.
We have now defined several functional domains of UNC80, including a NALCN-interacting domain, an UNC79-interacting domain, a soma-retention domain that restricts proteins in soma and a soma-retention relieve domain that overcomes soma retention and drives UNC80 toward neuronal processes including axon and dendrites.

UNC79 directly interacts with UNC80. In animals with UNC79 knocked out, the level of UNC80 is significantly reduced. The working mechanism of UNC79 is less understood.

The NALCN complex in mammalian brains contains the pore-forming subunit NALCN (~ 1500 amino acid (aa)) and at least two auxiliary subunits, UNC80 (~3,000 aa) and UNC79 (~3,000 aa). UNC80 serves as a “bridge” between NALCN and UNC79 by interacting with both. Neuropeptides (e.g. substance P, SP) and extracellular Ca2+ both regulate NALCN through G-protein coupled receptors (GPCRs) but with different downstream intracellular pathways: one is dependent on G proteins, but the other is G protein-independent and uses Src kinases (SFKs) instead.
Regulation of Neuronal Excitability by Extracellular Calcium via NALCN in a G-protein Dependent Pathway
Extracellular Ca2+ concentration ([Ca2+]e) is maintained at ~1.2 mM throughout the body, but significant fluctuation in [Ca2+]e exists in certain brain areas at different time of the day and during physiological conditions such as increased neuronal activities and pathological conditions such as seizures and brain ischemia. Extracellular Ca2+ regulates intrinsic neuronal excitability in a “negative” manner: a moderate decrease in [Ca2+]e usually excites neurons. The mechanism is generally believed to be via the interaction of Ca2+ ions with negative charges on the cell membrane (surface charge screening effects). We have discovered a novel mechanism (Lu et al. 2007): extracellular Ca2+ controls the sizes of NALCN-mediated Na+-leak, via a G-protein-dependent mechanism that requires the Ca2+-sensing receptor (CaSR) and UNC80. When NALCN or UNC79 is knocked out, decrease in [Ca2+]e from the physiological levels to sub-millimolar no longer excites neurons. Thus, NALCN contributes to the regulation of basal excitability by two major extracellular ions (Na+ and Ca2+).
Regulation of Neuronal Excitability by Neuropeptides via NALCN in a G-protein Coupled Receptor-dependent but G-protein -independent Pathway
Numerous neuropeptides regulate physiological processes such as feeding, rewarding, pain sensation, arousal and wakefulness. We are interested in how several neuropeptides such as substance P (SP) and neurotensin (NT) influence the properties of neurons. We discovered that SP and NT activate the NALCN complex. Along this line, we discovered a novel ion channel activation mechanism by G-protein coupled receptors: it is independent of G-protein but requires the Src family of tyrosine kinases and UNC80 (Lu et al. (2009, 2010)). The NALCN-UNC80 pathway appears to be a major mean by which t
he neuropeptides excite neurons: we and others have found that SP no longer excites neurons in several brain regions and spinal cord when NALCN is knocked out.
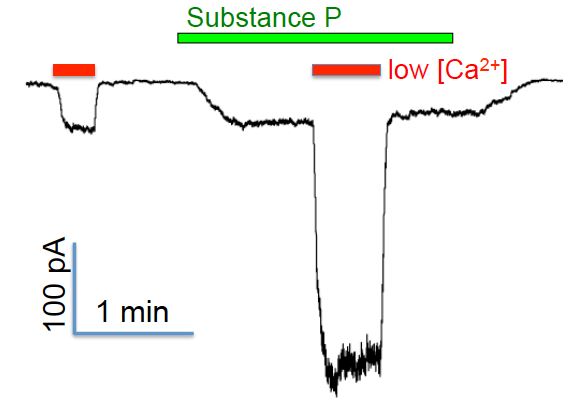
Synergistic regulation of NALCN by Ca2+ and substance P: Na+ leak currents recorded from a hippocampal neuron using voltage-clamp
Both the regulations of NALCN by extracellular Ca2+ and by substance P require UNC80. The two G-protein-independent and G-protein-dependent branches of NALCN regulation pathways are also synergistic: simultaneous lowering extracellular Ca2+ concentration and applying substance P leads to a sustained excitation to neurons much larger than the summation of those when the two stimuli are applied separately. Current efforts in this project focus on how receptor activation is coupled to channel opening and how such signaling events contribute to the intrinsic properties of individual neurons and associated neural circuit.
Physiological Roles of NALCN in Animal Behavior
A major function of NALCN-mediated Na+ “leak” appears to be in the generation of rhythmic behavior. All animals display long-period rhythmic behaviors such as circadian rhythm (~ 24 hours), as well as ones with shorter periods such as locomotion, heart beating, and breathing (milliseconds to seconds). We are interested in the molecular mechanisms underlying the generation and modulation of the “short-period” rhythms (Lu et al. 2007, Ren 2011). Using global and conditional knockout mouse models we developed, we and collaborators have discovered important roles of NALCN in the generation of rhythmic breathing patterns (Lu et al. 2007), the circadian rhythmic cycles of excitability of suprachiasmatic nucleus neurons (Flourakis et al. 2015) and the pace-making controlling the rhythmic movements of the gastrointestinal muscles (Kim et al. 2012).
NALCN- and UNC80- associated Human Diseases (link)
Inherited and spontaneous mutations in Nalcn gene in humans cause devastating symptoms including hypotonia, developmental delay, inability to sit, stand or walk, severe intellectual disability, epilepsy, lack of speech development and premature death in children. Using biochemical and electrophysiology methods, we study the consequences of the mutations to the biophysical and physiological properties of the channel. Working with clinical collaborators, we use mutant mouse lines to model the diseases resulting from mutations in genes encoding the NALCN complex and hopefully can use them for pilot treatment tests. To this end, our Nalcn and Unc79 mutant mouse models replicate nicely many of the key symptoms of human patients, including hypotonia, inability to stand, defects in rhythmic behavior, severe deficiency in cognitive function and premature death. The ultimate goal of this research is to find treatments or ways to relieve the symptoms in the patients.

Left: Nalcn and Unc79 knockout (-/-) mice are unable to stand or move, and have severe apnea (see Lu et al. 2007). Conditional knockout of Nalcn in forebrain causes severe deficiency in cognitive function (tested using fear conditioning learning and memory; unpublished). Similar symptoms have also been observed in human patients with NALCN mutations.
Key References:
Wie, J., Bharthur, A., Wolfgang, M., Narayanan, V., Ramsey, K., C4RCD Research Group, Kimberly Aranda, Zhang, Q., Zhou, Y. and Ren, D. (2020) Intellectual Disability-Associated UNC80 Mutations Reveal Inter-subunit Interaction and Dendritic Function of the NALCN Channel Complex. Nature Commun. (in press).
Flourakis, M., Kula-Eversole, E., Hutchison, A.L., Han, T.H, Aranda, K., Moose, D.L., White, K.P., Dinner, A.R., Lear, B.C., Ren, D, Diekman, C.D., Raman, I.M., and Allada, R. (2015) A Conserved bicycle model for circadian clock control of membrane excitability. Cell 162:836-848
Lu B, Zhang Q, Wang H, Wang Y, Nakayama M, Ren D (2010) Extracellular Calcium Controls Background Current and Neuronal Excitability via an UNC79-UNC80-NALCN Cation Channel Complex. Neuron 68:488-499
Lu, B.*, Su, Y.*, Das, S., Wang, H., Wang, Y., Liu, J. and Ren, D. (2009) Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature 457:741-744
Lu, B., Su, Y., Das, S., Xia, J., Liu, J. and Ren, D. (2007) The neuronal NALCN channel contributes resting sodium permeability and is required for normal respiratory rhythm. Cell 129: 371-378
Ren, D. (2011) Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron 72: 899-911